Steven U. Walkley, D.V.M., Ph.D.
Professor, Dominick P. Purpura Department of Neuroscience
Professor, Department of Pathology
Professor, The Saul R. Korey Department of Neurology
Director, Rose F. Kennedy Intellectual and Developmental Disabilities Research Center, Dominick P. Purpura Department of Neuroscience
(neuroscience category)

Pathobiology and treatment of genetic brain diseases
Training in Comparative Medicine and Neuroscience provided the basis for my career interests in neurogenetic disease, particularly those disorders impacting neuronal homeostatic mechanisms and resulting in intellectual disability and related neurobehavioral abnormalities. My lab has published extensively in the area of pathogenic cascade analysis in lysosomal disease, defining key changes in neuronal structure and function as a consequence of lysosomal compromise. Current studies include: (i) the causes and consequences of ectopic dendritogenesis and neuroaxonal dystrophy, (ii) altered synaptic function underlying intellectual compromise, (iii) involvement of mTOR and TFEB/TFE3 in homeostatic dysregulation following lysosomal compromise and its impact on endosomal and autophagasomal function, and (iv) the importance of metabolite salvage in lysosomal processing.
Diseases of current focus include the lysosomal diseases Niemann-Pick types A and C, mucolipidosis IV, cystinosis, GM1 and GM2 gangliosidosis, Sanfilippo type A (MPS IIIA), Batten disorders (CLN2 and CLN3) and a newly discovered endosomal disorder known as Christianson syndrome.
My lab is also significantly involved in therapy development for genetic brain disease. We were the first to show essentially complete correction of CNS disease in the lysosomal disorder known as alpha-mannosidosis through the use of bone marrow transplantation and this treatment approach is now the standard of care for children diagnosed with this rare disorder. A disease of current focus toward therapy is Niemann-Pick type C (NPC), a fatal cholesterol-glycosphingolipid lysosomal storage disorder of children. Based on our studies of glycosphingolipid processing abnormalities in NPC disease we developed the first and presently only approved (by EMEA; FDA pending) therapy for this disorder. This is the imino sugar known as N-butyldeoxynojirimycin, or miglustat, which is a partial inhibitor of glycosphingolipid synthesis.
More recently we discovered that the FDA-approved excipient known as hydroxypropyl beta-cyclodextrin is efficacious in limiting intraneuronal accumulation of both unesterified cholesterol and glycosphingolipids, and dramatically extends the lifespan in animal models of NPC disease. Research and clinical trial development was subsequently pursued through a unique scientist/clinician/parent consortium known as SOAR (Support Of Accelerated Research) for NPC disease (SOAR-NPC) and through collaboration with TRND (Therapeutics for Rare and Neglected Diseases) and NCATS (National Center for Advancing Translational Sciences) at NIH. Phase 1 trials of this compound in NPC patients began in February, 2013, followed by the Phase 2/3 trial, with outcome analysis now in progress.
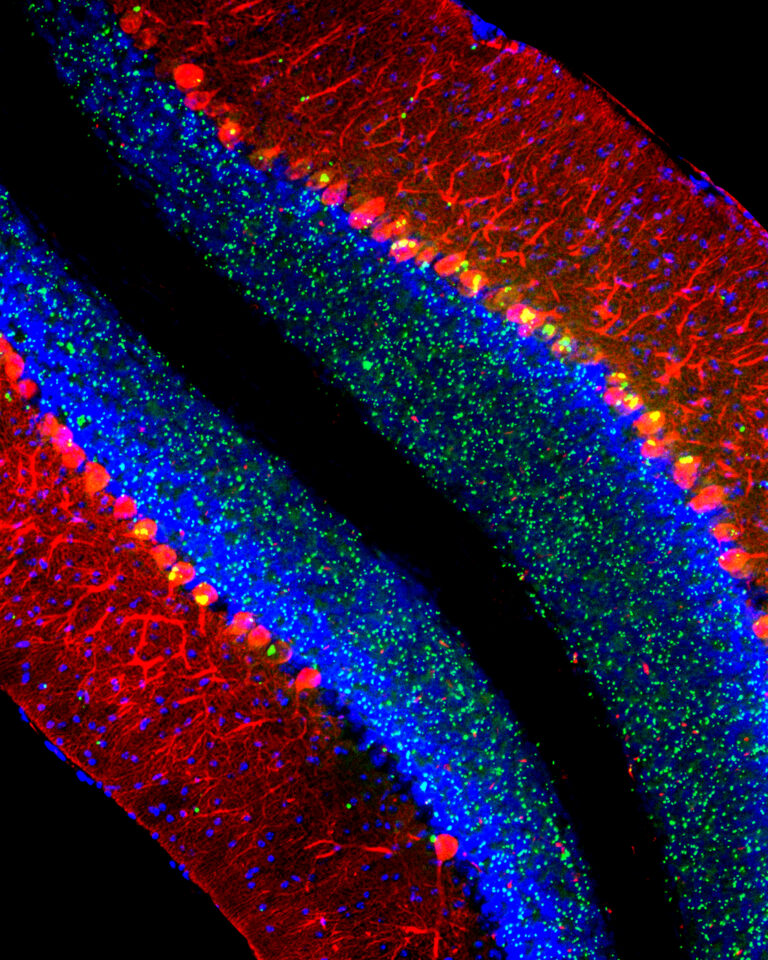
“Cerebellum of a mouse modeling the lysosomal disease late-infantile neuronal ceroid lipofuscinosis (CLN2 disease). Purkinje cells are labeled red, nuclei are blue, and aberrant aggregates of the protein p62/Sqstm1 are green.” (Matt Micsenyi and Steve Walkley)
Selected Publications
Walkley SU, Abbeduto L, Batshaw ML, Bhattacharyya A, Bookheimer SY, Christian BT, Constantino JN, de Vellis J, Doherty DA, Nelson DL, Piven J, Poduri A, Pomeroy SL, Samaco RC, Zoghbi HY, Guralnick MJ; IDDRC Directors Committee. Intellectual and Developmental Disabilities Research Centers: 50 Years of Scientific Accomplishments, Ann Neurol. 2019 Jun 17. doi: 10.1002/ana.25531. [Epub ahead of print] Review. PMID:31206741
Vite, CH, Bagel, JH, Swain, GP, Prociuk M, Sikora, J… and Walkey, SU. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type CI disease. Sci Transl Med 2015 Feb 25;7(276):276ra26.doi:10.1126/ scitranslmed. 3010101 PMID: 25717099 PMCID: PMC4415615
Micsenyi MC, Sikora J, Stephney G, Dobrenis K, Walkley SU. Lysosomal membrane permeability stimulates protein aggregate formation in neurons of a lysosomal disease. J Neurosci 33:10815–10827, 2013. PMID: 23804102 [PubMed – in process] PMCID: PMC3693058
Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8(5):719–30. Epub 2012/06/01. doi: 10.4161/auto.19469. PMID: 22647656; PMCID: PMC3378416.
Strømme P, Dobrenis K, Sillitoe RV, Gulinello M, Ali NF, Davidson C, Micsenyi MC, Stephney G, Ellevog L, Klungland A, Walkley SU. X-linked Angelman-like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal-lysosomal dysfunction. Brain : a journal of neurology. 2011;134(Pt 11):3369–83. Epub 2011/10/04. doi: 10.1093/brain/awr250. PMID: 21964919; PMCID: PMC3212719.
McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. The Journal of comparative neurology. 2004;480(4):415–26. Epub 2004/11/24. doi: 10.1002/cne.20355. PMID: 15558784.
Walkley SU. Pathogenic cascades in lysosomal disease-Why so complex? Journal of inherited metabolic disease. 2009;32(2):181–9. Epub 2009/01/09. doi: 10.1007/s10545-008-1040-5. PMID: 19130290; PMCID: PMC2682782.
Davidson CD, Ali NF, Micsenyi MC, Stephney G, Reanult S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PloS one. 2009;4(9):e6951. Epub 2009/09/15. doi: 10.1371/journal.pone.0006951. PMID: 19750228; PMCID: PMC2736622.
Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Current biology : CB. 2001;11(16):1283–7. Epub 2001/08/30. PMID: 11525744.
Walkley SU, Thrall MA, Dobrenis K, Huang M, March PA, Siegel DA, et al. Bone marrow transplantation corrects the enzyme defect in neurons of the central nervous system in a lysosomal storage disease. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(8):2970–4. Epub 1994/04/12. PMID: 8159689; PMCID: PMC43496.