R. Suzanne Zukin, Ph.D.
Professor, Dominick P. Purpura Department of Neuroscience
F.M. Kirby Chair of Neural Repair and Protection
Director, Neuropsychopharmacology Center
(neuroscience category)

Regulation of synaptic function and plasticity in response to external cues including neuronal insults, maternal deprivation, and stress, via epigenetic mechanisms. Altered signaling at the synapse in mouse models of autism.
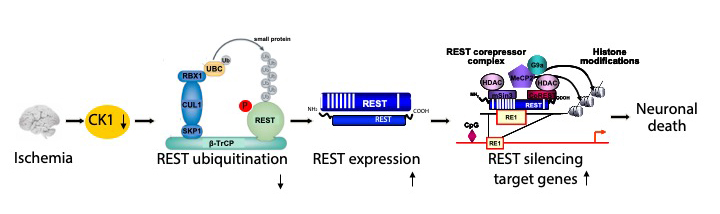
There are four major lines of ongoing research in the Zukin lab. First, we are studying the molecular and cellular mechanisms that regulate N-methyl-D-aspartate-type glutamate receptor (NMDA receptor) expression at synapses in the brain. We discovered that the switch in NMDA receptor phenotype at hippocampal synapses during normal brain development is regulated by epigenetics in an experience-dependent manner. In normal brain, the gene silencing transcription factor REST is activated during a brief window of time in differentiated neurons of the hippocampus, a brain center implicated in learning and memory, and drives the switch from immature to mature NMDA receptors. Remarkably, depriving pups of maternal access for brief periods of time during the first postnatal week prevents activation of REST and epigenetic modifications essential to acquisition of mature NMDA receptors and normal brain development. These findings have striking implications for treatment of anxiety, post-traumatic stress and other disorders associated with early maternal separation. New questions are: What is the mechanism by which REST is activated during brain development? Do other forms of stress regulate the switch in NMDA receptors? What are the consequences of blocking the switch? Our interest stems from the fact that NMDA receptors play a central role in cognitive functions such as learning and memory, synaptic plasticity and formation of neural circuitry. NMDA receptor dysregulation is implicated in Alzheimer’s disease, Huntington’s disease, AIDS dementia, stroke and schizophrenia.
Second, we are studying the molecular and cellular mechanisms that underlie the neuronal death associated with stroke and epilepsy. We discovered that neuronal insults activate REST in selectively vulnerable adult hippocampal neurons. Upon activation, REST orchestrates epigenetic reprogramming of neuronal genes in differentiated neurons. We further showed that prolonged activation of REST is causally related to neuronal death in a clinically-relevant model of ischemic stroke. A key downstream target of REST in insulted CA1 neurons is the gene encoding the AMPA receptor subunit GluA2. This is of interest because the GluA2 subunit governs calcium permeability, channel conductance and AMPA receptor trafficking to and from synaptic sites. GluA2-lacking AMPA receptors are highly permeable to calcium and zinc, which rise to toxic levels in insulted neurons. Objectives are: 1) to understand how REST is activated in insulted neurons; 2) to examine epigenome-wide dysregulation of REST targets in stroke, Huntington’s disease and Alzheimer’s disease; and 3) to identify novel strategies to protect the human brain from neurodegeneration. Our interest stems from the known role of AMPA receptors in neuronal death arising in stroke, epilepsy, ALS and spinal cord injury.
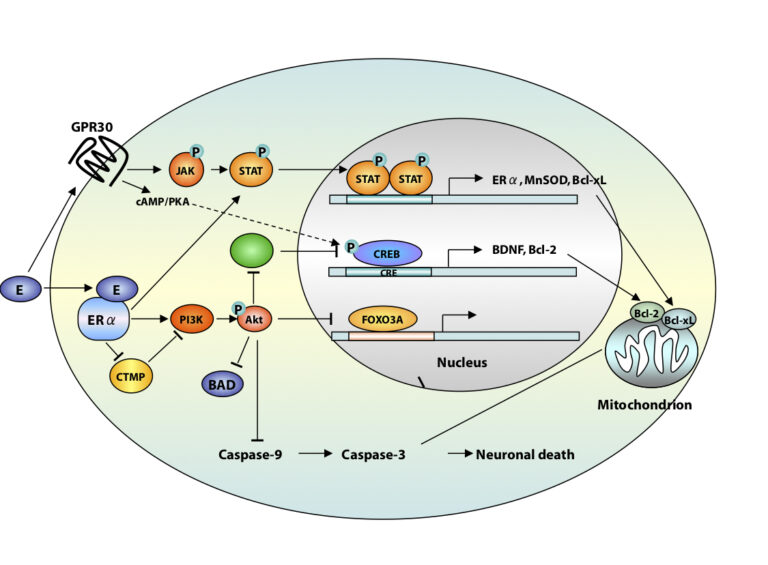
A third area of interest is that of estrogen neuroprotection in animal models of stroke, including global ischemia. Recently, we found that long-term treatment with estrogen at physiological levels ameliorates death of hippocampal neurons and cognitive deficits associated with global ischemia. We showed that ischemia and estrogen act synergistically to activate the transcription factor STAT3 and promote transcription of survivin, an inhibitor of apoptosis protein and gene target of STAT3, in insulted CA1 neurons. In experiments in which we employ direct delivery of shRNA constructs into the hippocampal CA1 of living animals, we found that STAT3 and survivin are essential to estrogen neuroprotection. These findings identify STAT3 and survivin as therapeutic targets in a clinically-relevant model of stroke. Objectives are to identify epigenetic mechanisms by which estrogen rescues neurons. Our interest stems from data that estrogen reduces the risk of cardiac arrest and stroke in animal models.
A fourth area of interest is that of RNA trafficking and targeting to dendrites and local protein synthesis in Fragile X syndrome. We found that mTOR signaling is overactivated in hippocampal neurons of Fragile X mice and causally related to aberrant synaptic plasticity. We also found that targeting of AMPAR mRNAs to synapses under basal conditions and in response to mGluR signaling is dysregulated in Fragile X neurons. We are using a combination of high resolution imaging of individual mRNA molecules (in collaboration with the Singer lab), molecular biology, and electrophysiology to examine AMPAR mRNA trafficking, local translation, synaptic plasticity and spine structure in Fragile X mice. Objectives are to identify novel signaling pathways that play a role in synaptic dysfunction. We believe that understanding the mechanisms responsible for abnormal function at the synapse will advance novel therapeutic strategies to ameliorate cognitive deficits in Fragile X syndrome and unlock doors for treating other autism spectrum disorders.
Positions for graduate students and post-doctoral fellows are available in all four areas of the laboratory’s research. Independent researchers and ideas are welcome, while well-defined and achievable projects are waiting for motivated, young investigators.
Selected Publications
(From a total of 194 peer-reviewed papers and 41 book chapters)
Gompers A, Hwang J-Y, Monday HR, Buxbaum A, Yan J, Sawicka K, Castillo PE, Singer RH, Zukin RS The memory protein CPEB3 reduces synaptic incorporation of Ca2+-permeable AMPARs and loss of anti-Hebbian LTP at synapses onto inhibitory interneurons in Fragile X mice. Nat Neurosci, in review, 2017.
Yan J, Porch WM, Court-Vazquez B, Bennett MV, Zukin RS Activation of autophagy rescues synaptic and cognitive deficits in Fragile X mice Proc. Natl. Acad. Sci. USA, 2018. PMID:30242133.
Hwang JY, Zukin RS REST, a master transcriptional regulator in neurodegenerative disease. Curr Opin Neurobiol. 48:193–200, 2018. PMID:29351877.
Pyronneau A, Qionger H, Hwang J-Y, Contractor A, Zukin RS Enhanced Rac1/cofilin signaling is critical to dendritic spine defects, synaptic dysfunction and impaired sensory perception in Fragile X Syndrome. Sci Signal 10: pii: eaan0852, 2017. PMID:29114038.
Hwang J-Y, Aromolaran KA, Zukin RS The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat Rev Neurosci 18:347–361, 2017. PMID: 28515491
Hwang J-Y, Gertner MJ, Pontarelli F, Bennett MVL, Ofengeim D, Zukin RS Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ 24:317–329, 2017. PMID: 27935582
Sawicka K, Pyronneau A, Chao M, Bennett MV, Zukin RS Elevated ERK/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in fragile X mice. Proc Natl Acad Sci USA 113:E6290–E6297, 2016. PMID: 27663742
Choi CH et al., Multiple drug treatments that increase cAMP signaling restore long-term memory and aberrant signaling in Fragile X syndrome models. Front Behav Neurosci 10:136–142. 2016. PMID:27445731
Puckerin A, Aromolaran KA, Chang DD, Zukin RS, Colecraft HM, Boutjdir M, Aromolaran AS. hERG 1a LQT2 C-terminus truncation mutants display hERG 1b-dependent dominant negative mechanisms. Heart Rhythm. 13:1121–30, 2016. PMID: 26775140
Huber KM, Klann E, Costa-Mattioli M, Zukin RS. Dysregulation of Mammalian Target of Rapamycin Signaling in Mouse Models of Autism. J Neurosci. 35:13836–42, 2015. PMID: 26468183